Amphotericin B
It was orginally extracted from a bacterium Streptomyces nodosus in 1955 at the Squibb Institute of Medical Research. This compound is called amphotericin B and now it is listed on the World Health Organisation's List of Essential Medicine, which is the list of the most important medications needed in basic health. Amphotericin B works as antifungal drug and is often used intravenously for systemic fungal infections. The name of amphotericin B is derived from its amphoteric property.
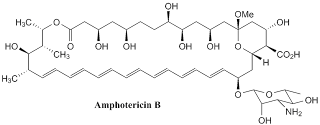
Amphotericin B is one of the member of the macrolide family of natural products and one of sub-family of macrolide family is polyene family. This sub-family, including amphotericin B, poses formidable challenges to synthetic organic chemistry. In 1987, Nicolaou's group made a breakthrough for being the first group to synthesis this polyene macrolide. In general, the synthesis can be described into 2 phases: synthesis of protected amphoteronolide B and glycosidation.
 The first stage of the synthesis of amphotericin B by Nicolaou's group was the formation of the first fragment of amphoteronolide B which serves as ketophosphonate 8. The starting material of 8 was the enantiomeric pair of xylose which underwent acetonide formation. This acetonide protects 1- and 2-OH, which allowed 3-OH to be eliminated giving 3-deoxy compound. (+)-Xylose was transformed into an aldehyde 2a while (-)-xylose formed the second fragment 2b which has ketophosphonate group. Both compounds were connected via Horner-Wadsworth-Emmons reaction followed by hydrogenation and then transformed into ketophosphonate. In this phase, ketophosphonate 8 was formed with the OH groups are protected by either acetonide or TBS.
The first stage of the synthesis of amphotericin B by Nicolaou's group was the formation of the first fragment of amphoteronolide B which serves as ketophosphonate 8. The starting material of 8 was the enantiomeric pair of xylose which underwent acetonide formation. This acetonide protects 1- and 2-OH, which allowed 3-OH to be eliminated giving 3-deoxy compound. (+)-Xylose was transformed into an aldehyde 2a while (-)-xylose formed the second fragment 2b which has ketophosphonate group. Both compounds were connected via Horner-Wadsworth-Emmons reaction followed by hydrogenation and then transformed into ketophosphonate. In this phase, ketophosphonate 8 was formed with the OH groups are protected by either acetonide or TBS.
 In the second stage, the highly conjugated polyene-aldehyde was formed. Again, in this step ketophosphonate-aldehyde condensation is the key reaction to control the stereochemistry of the polyene component of 21. This compound was formed via this condensation twice with the aldehyde and phosphonate 19. The aldehyde was prepared via Evans aldol followed by several steps of reduction and deprotection. It is also noteworthy that the reagent 5 that was used in the first step was also synthesised from the same starting material which is (+)-diethyl-L-tartrate. Another interesting point to note, an intermediate compound that is not quite related to 21 such as epoxide 11 that is formed under Sharpless condition was used.
In the second stage, the highly conjugated polyene-aldehyde was formed. Again, in this step ketophosphonate-aldehyde condensation is the key reaction to control the stereochemistry of the polyene component of 21. This compound was formed via this condensation twice with the aldehyde and phosphonate 19. The aldehyde was prepared via Evans aldol followed by several steps of reduction and deprotection. It is also noteworthy that the reagent 5 that was used in the first step was also synthesised from the same starting material which is (+)-diethyl-L-tartrate. Another interesting point to note, an intermediate compound that is not quite related to 21 such as epoxide 11 that is formed under Sharpless condition was used.
 Joining two fragments 8 and 21 was done via esterification using DCC and DMAP and then followed by ketophosphonate condensation macrocyclisation under the base of K2CO3. It is interesting the use of weaker base K2CO3 in the presence of [18]crown-6 rather than using stronger LDA as in the previous stages. The crown ether enhances the basicity of K2CO3 due unsolvated base. The glycosidation poses a problem which is the requirement for a β-glycoside bond in a 1,2-cis relationship with C2 hydroxyl group of the carbohydrate unit. This formidable challenge was addressed by construction of mycosamine equivalent 24. The glycosidation occurs with the aid of acid PPTS catalyst. The final steps of this total synthesis were deprotection and reduction of azide via dithiol and triethylamine.
Joining two fragments 8 and 21 was done via esterification using DCC and DMAP and then followed by ketophosphonate condensation macrocyclisation under the base of K2CO3. It is interesting the use of weaker base K2CO3 in the presence of [18]crown-6 rather than using stronger LDA as in the previous stages. The crown ether enhances the basicity of K2CO3 due unsolvated base. The glycosidation poses a problem which is the requirement for a β-glycoside bond in a 1,2-cis relationship with C2 hydroxyl group of the carbohydrate unit. This formidable challenge was addressed by construction of mycosamine equivalent 24. The glycosidation occurs with the aid of acid PPTS catalyst. The final steps of this total synthesis were deprotection and reduction of azide via dithiol and triethylamine.

The total synthesis of amphotericin B shows a high demand of ketophosphonate reaction and Horner-Wadsworth-Emmons reaction for the construction of C=C bond as it was used 5 times and also the intramolecular ketophosphonate reaction for the construction of 38-membered ring.
Amphotericin B works as antifungal drug via binding with ergosterol, a component of fungal cell membranes, forming a transmembrane channel that leads to monovalent ion leakage. However, the actual mechanism may be more complex.
 Amphotericin B is only used clinically because it is significantly more active in vivo. Amphotericin A, the brother amphotericin B, is almost identical but has little antifungal activity.
Amphotericin B is only used clinically because it is significantly more active in vivo. Amphotericin A, the brother amphotericin B, is almost identical but has little antifungal activity.
References

Amphotericin B is one of the member of the macrolide family of natural products and one of sub-family of macrolide family is polyene family. This sub-family, including amphotericin B, poses formidable challenges to synthetic organic chemistry. In 1987, Nicolaou's group made a breakthrough for being the first group to synthesis this polyene macrolide. In general, the synthesis can be described into 2 phases: synthesis of protected amphoteronolide B and glycosidation.




The total synthesis of amphotericin B shows a high demand of ketophosphonate reaction and Horner-Wadsworth-Emmons reaction for the construction of C=C bond as it was used 5 times and also the intramolecular ketophosphonate reaction for the construction of 38-membered ring.
Amphotericin B works as antifungal drug via binding with ergosterol, a component of fungal cell membranes, forming a transmembrane channel that leads to monovalent ion leakage. However, the actual mechanism may be more complex.

References
- K. C. Nicolaou, R. A. Daines, J. Uenishi, W. S. Li, D. P. Papahatjis, and T. K. Chakrabothy, J. Am. Chem. Soc., 1987, 109, 2205.
- K. C. Nicolaou, R. A. Daines, T. K. Chakrabothy, and Y. Ogawa, J. Am. Chem. Soc., 1987, 109, 2821.
- K. C. Nicolaou, R. A. Daines, T. K. Chakrabothy, and Y. Ogawa, J. Am. Chem. Soc., 1988, 110, 4685.
Comments